
Besoin de commercialiser votre dispositif médical aux Etats Unis ? Alors vous devrez obtenir l’agrément FDA ! On vous dit tout.
L’agrément FDA, également connu sous le nom d’approbation de la Food and Drug Administration (FDA), fait référence à l’autorisation accordée par la FDA des États-Unis pour la commercialisation et la distribution d’un produit réglementé, notamment les dispositifs médicaux, les médicaments, les produits biologiques, les aliments, les compléments alimentaires, et autres.
Pour être commercialisés aux États-Unis, tous les dispositifs médicaux doivent être enregistrés auprès de la Food and Drug Administration (FDA). Le processus de mise sur le marché des dispositifs médicaux aux États-Unis peut être ardu, impliquant ainsi une classification, une préparation minutieuse, et la soumission d’une demande à la Food and Drug Administration (FDA).
L’objectif de cet agrément : garantir que les produits réglementés répondent à des normes strictes de sécurité, d’efficacité et de qualité avant d’être mis sur le marché américain. Les exigences de la FDA pour la mise sur le marché d’un dispositif médical aux États-Unis sont nombreuses. Les entreprises désirant commercialiser leurs dispositifs médicaux sur le marché américain doivent suivre quelques étapes :
Classification du dispositif :
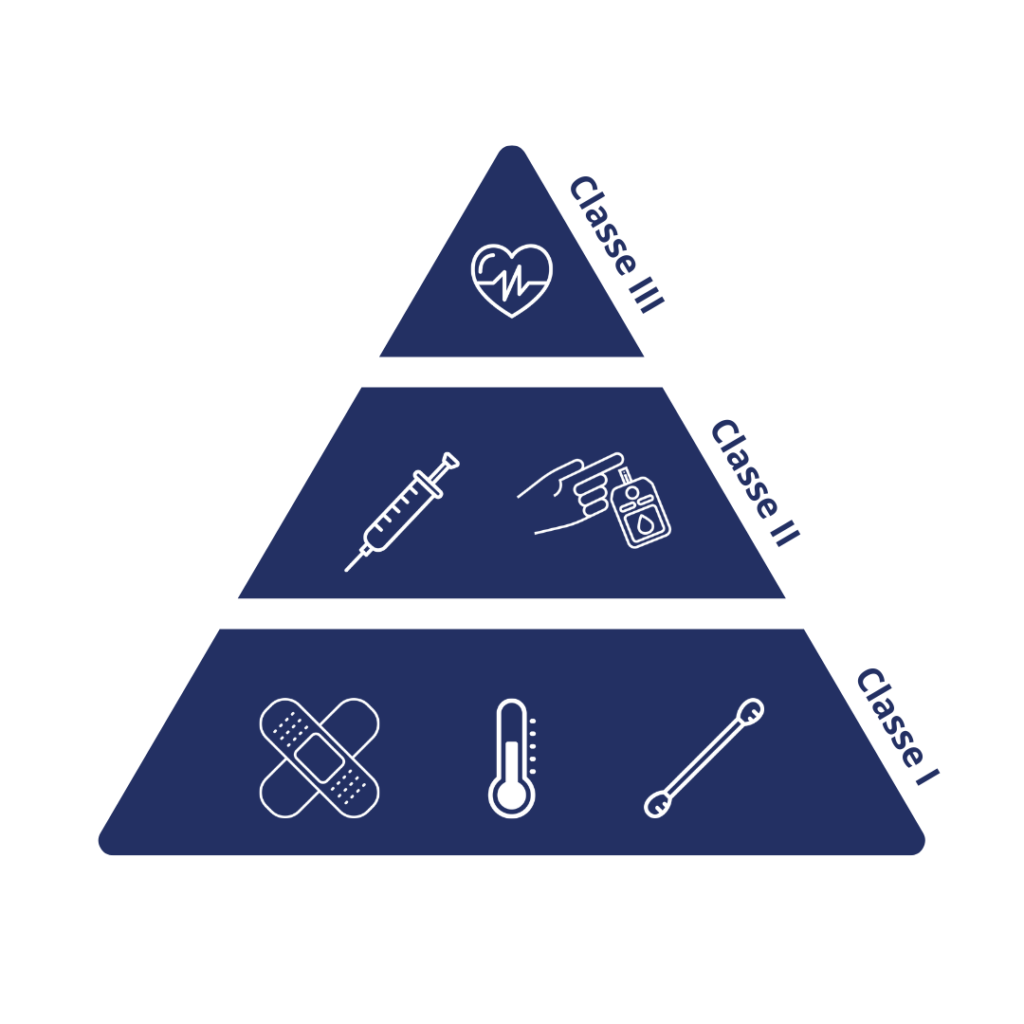
La première étape consiste à classifier le dispositif en fonction de son risque pour le patient/utilisateur, de son utilisation prévue et de ses indications d’utilisation.
Les dispositifs sont divisés en trois classes :
Classe I : Les dispositifs de Classe I sont ceux qui présentent le plus faible niveau de risque pour les utilisateurs. Ils sont généralement simples et ne nécessitent pas de contrôle strict. Exemples : pansements, thermomètres, coton, etc.
Classe II : Présentant un risque modéré et nécessitant des contrôles spéciaux en plus des contrôles généraux. Exemples : seringues, glucomètres, etc.
Classe III : Les dispositifs de Classe III sont ceux qui présentent le plus haut niveau de risque pour les utilisateurs ou sont utilisés dans des situations critiques. Ils sont soumis à la réglementation la plus stricte de la FDA et nécessitent généralement des études cliniques approfondies pour démontrer leur sécurité et leur efficacité avant d’être approuvés. Exemples : stimulateurs cardiaques implantables, implants mammaires, systèmes de diagnostic in vitro, etc.
Identification de la demande de pré commercialisation :
Chaque classe de dispositif médical nécessite une soumission de pré commercialisation spécifique, comprenant :
– Notification de pré commercialisation 510(k) pour les dispositifs “substantiellement équivalents” à un dispositif déjà commercialisé aux États-Unis.
– Demande d’approbation préalable à la mise sur le marché (Premarket Approval Application – PMA) pour les dispositifs de Classe III, nécessitant des preuves scientifiques valides démontrant leur sécurité et leur efficacité.
– Évaluation pour les nouveaux dispositifs de risque faible à modéré (Classe I ou II) lorsque aucun dispositif équivalent n’est identifié.
– Exemption pour dispositifs humanitaires (HUD) pour les dispositifs de Classe III traitant des maladies ou infections rares.
Identification de la demande de pré commercialisation :
Chaque classe de dispositif médical nécessite une soumission de pré commercialisation spécifique, comprenant :
– Notification de pré commercialisation 510(k) pour les dispositifs “substantiellement équivalents” à un dispositif déjà commercialisé aux États-Unis.
– Demande d’approbation préalable à la mise sur le marché (Premarket Approval Application – PMA) pour les dispositifs de Classe III, nécessitant des preuves scientifiques valides démontrant leur sécurité et leur efficacité.
– Évaluation pour les nouveaux dispositifs de risque faible à modéré (Classe I ou II) lorsque aucun dispositif équivalent n’est identifié.
– Exemption pour dispositifs humanitaires (HUD) pour les dispositifs de Classe III traitant des maladies ou infections rares.
Préparation de la soumission pré commercialisation :
Pour préparer correctement la demande, il est essentiel d’inclure toutes les informations pertinentes, telles que les contrôles de conception, les tests non cliniques, les tests cliniques et l’étiquetage, en fonction de la classification du dispositif.
Soumission de la demande à la FDA :
Il faut ensuite soumettre la demande à la FDA en suivant les procédures et les exigences spécifiques à la voie choisie. Il est important d’inclure tous les documents requis et de payer les frais applicables. La FDA examinera la demande pour déterminer sa complétude, avec des délais de réponse variant de 90 à 180 jours selon la classe du dispositif et le type de demande.
Enregistrement de l’entreprise et du dispositif :
L’entreprise doit effectuer un enregistrement annuel auprès de la FDA et référencer en ligne les dispositifs commercialisés. Le marché des dispositifs médicaux aux États-Unis offre de vastes opportunités aux fabricants, à condition de satisfaire à ces nombreuses exigences. Une compréhension approfondie du processus peut accélérer la mise sur le marché des produits.
Commercialisation et suivi post commercialisation :
Une fois que le dispositif a été approuvé par la FDA, il peut être commercialisé aux États-Unis. Il faut ensuite s’assurer de suivre toutes les exigences de surveillance post-commercialisation, telles que le suivi des effets indésirables et les rapports réguliers à la FDA.
En résumé, l’agrément FDA est une autorisation délivrée par la Food and Drug Administration des États-Unis pour la commercialisation de produits réglementés, et il garantit que ces produits répondent à des normes strictes de sécurité, d’efficacité et de qualité.
Il est important de noter que les exigences spécifiques peuvent varier en fonction du type de dispositif et des circonstances individuelles.



{kind=link}